The Baby, the Base Editor, and the Breakthrough: How Scientists Fixed a Gene in Just Six Months
May 25, 2025
Discover how a custom CRISPR base-editing therapy saved an infant with a rare genetic disorder-and why affordable, accessible genetic testing is now more critical than ever.
In the summer of 2024, a baby named KJ Muldoon entered the world under a silent threat. Within two days of life, his tiny body was overwhelmed by ammonia-a toxic byproduct of protein digestion-spiking to levels that could cause brain damage, coma, or death.
The diagnosis was swift but devastating: carbamoyl-phosphate synthetase 1 deficiency, or CPS1 deficiency, a rare genetic disorder that disables the body's main ammonia detox system. Doctors at Children’s Hospital of Philadelphia (CHOP) and University of Pennsylvania knew the odds: about 50% of babies with this condition don’t survive infancy.
But instead of accepting those odds, they tried something no one had ever done before.
They designed a custom gene editor-just for KJ-and infused it directly into his bloodstream.
It was the first in-human, patient-specific base editing therapy ever given in vivo. And it worked.
Here’s how this landmark moment in science unfolded-one base pair, one molecule, one miracle at a time.
The Culprit: A Single-Letter Genetic Error
KJ’s CPS1 deficiency was caused by two faulty copies of the CPS1 gene-one from each parent. This gene encodes an enzyme critical for the urea cycle, which turns toxic ammonia into harmless urea that we excrete in urine.
In KJ’s case, genome sequencing found two "nonsense mutations"-genetic typos that insert a premature stop signal into the gene:
- Q335X on the paternal allele: A C to T change that halts protein production early.
- E714X on the maternal allele: Another stop mutation.
Together, these errors rendered his liver incapable of clearing ammonia. Even specialized medications and protein-restricted diets could only delay the inevitable: a liver transplant or worse.
But there was a glimmer of opportunity: what if you could fix just one of those mutations?
The Plan: Rewrite the Mutation with Base Editing
CRISPR technology is now famous, but the early versions (CRISPR 1.0) worked by cutting both strands of DNA-a bit like smashing a window and hoping it reassembles in a better shape.
Instead, the team at CHOP and Penn used CRISPR 2.0, also known as base editing. This technology allows scientists to change a single DNA letter-like turning a typo into the correct word-without breaking the DNA.
The team chose to target the Q335X mutation (from Dad), which required converting a stop codon back to a functional amino acid by changing an adenine (A) to a guanine (G).
This is where the science becomes both astonishing and elegant.
The Custom Toolkit: Kayjayguran and Abengcemeran
Because this mutation was unique to KJ, the team had to custom-design the entire therapy:
- A Guide RNA (gRNA): A short strand of RNA that acts like a GPS, directing the editor to the exact mutation site.
They named it “Kayjayguran”, a nod to KJ. - An Adenine Base Editor (ABE): A CRISPR protein fused to an enzyme that converts A to G.
This version-NGC-ABE8e-V106W-was optimized for precision and minimal side effects. They named its mRNA “Abengcemeran.”
Together, this bespoke editor-packaged in lipid nanoparticles-became known as “k-abe.”
Within two months of KJ’s birth, the entire construct was ready for testing.
Preclinical Testing at Lightning Speed
Creating a drug from scratch in six months sounds reckless-unless you build in layers of validation.
The researchers raced to verify that the editor worked and was safe:
- In human liver cell lines (HuH-7) modified to carry KJ’s mutation
- In engineered mice with KJ’s exact genetic error
- In non-human primates, to check for immune responses and toxicity
They even performed off-target analysis using multiple state-of-the-art tools like ONE-seq, CHANGE-seq, and GUIDE-seq, to make sure the editor wasn’t hitting the wrong DNA sequences.
At every step, they refined the gRNA, confirmed the edit’s accuracy, and ruled out concerning bystander edits.
By month five, they had enough data to submit an emergency request to the FDA for compassionate use.
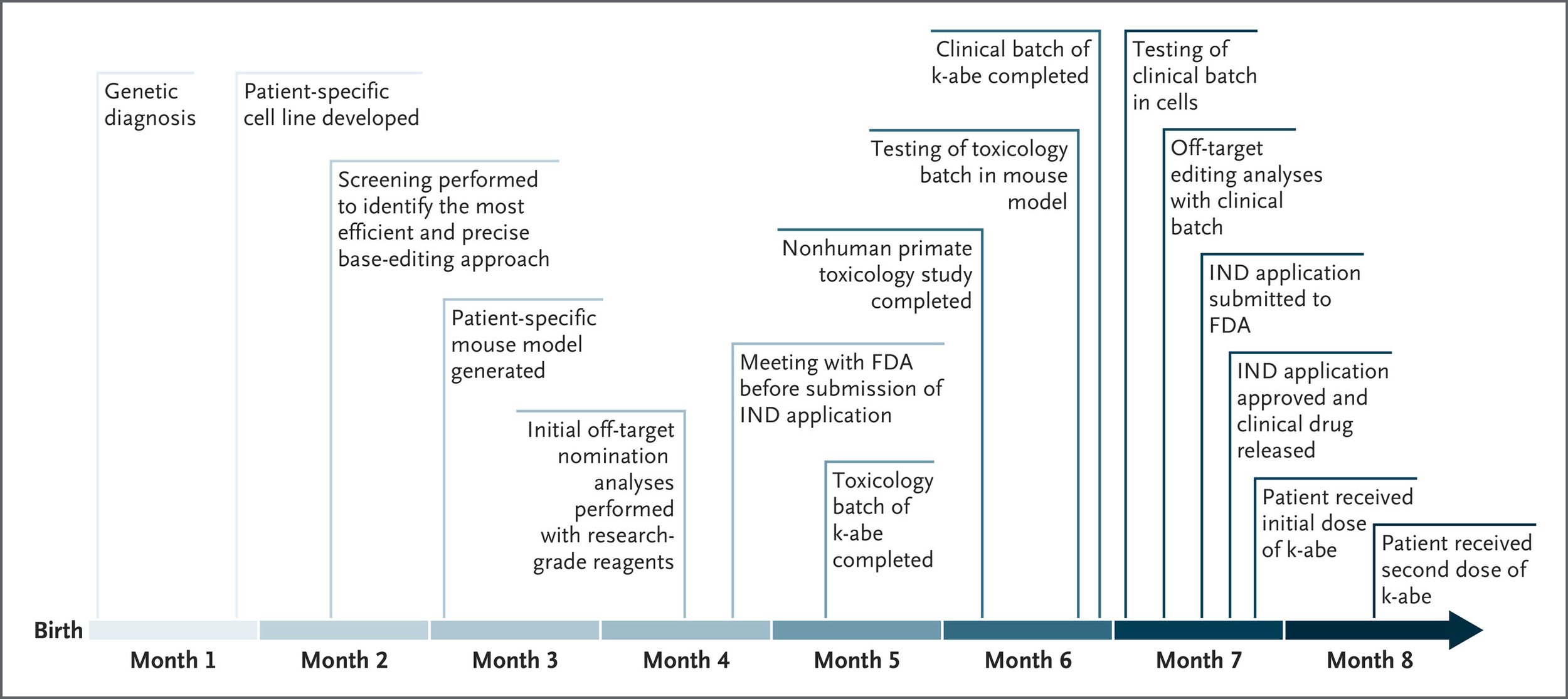
The Infusions: Editing a Gene in a Living Human
At 7 months old, KJ received his first infusion of k-abe, at a cautious low dose (0.1 mg/kg). No severe side effects occurred, and his protein intake could be gradually increased.
Three weeks later, he received a second, higher dose (0.3 mg/kg). This time, the signs of efficacy were clearer:
- Lower blood ammonia levels
- Reduction in medication by 50%
- No metabolic crises, even during two viral infections
- Normal developmental progress
A third dose was given in April 2025 (not yet reported in NEJM), and further monitoring is ongoing.
They didn’t perform a liver biopsy (too risky in a fragile infant), but based on his clinical response, the gene editing appears to be working-at least partially, and safely.
The Bigger Picture: Why This Is Just the Beginning
This wasn’t just a treatment. It was a template.
The researchers have shown that it’s now possible to:
- Sequence a patient
- Identify a disease-causing variant
- Design a base editor
- Test it in cells, mice, and primates
- Get regulatory approval
- Infuse it into a human
- Observe real-world benefit
- Do it all in six months
This has staggering implications-not only for rare diseases like CPS1 deficiency, but potentially for thousands of single-nucleotide conditions affecting the brain, lungs, muscles, and heart.
But Innovation Is Only Half the Story
Here’s the hard truth: none of this would have happened if KJ hadn’t received full genome sequencing within days of birth.
Most children with genetic disorders don’t.
Genetic testing is still too expensive, too limited, and often not covered by insurance.
Yet we now have the tools to correct a single typo in our DNA. It’s no longer science fiction-it’s clinical fact. But to unlock its full potential, we must ensure everyone can be diagnosed early and accurately.
This means investing in:
- Affordable, targeted mutation panels
- Routine sequencing for high-risk newborns
- Wider public insurance coverage for genetic testing
Without the diagnosis, there can be no cure.
Conclusion: A First Step Toward a New Era
KJ’s story is extraordinary-but it should not be rare. The science is ready. The tools exist. Now the question is whether we can build a healthcare system that makes personalized genetic medicine available to everyone who needs it.
Because the next KJ is already out there.
And they deserve to be found.
Sources:
- Musunuru, K., et al. (2025). Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. NEJM. DOI: 10.1056/NEJMoa2504747
- Topol, E. (2025). The First Human to Undergo In Vivo CRISPR 2.0 Personalized Genome Editing. Ground Truths, May 18, 2025.
Have questions about this research or our products?
Contact Our TeamGet new posts delivered to your inbox